Having a great medical IoT product isn’t enough to present it on the market that is strictly regulated worldwide — it has to be fully compliant with accepted standards for medical devices. Oftentimes, developers think about it during the later development stages and that can hold back the whole process and make them go back in time and do the same work twice. This approach is time-consuming, deadline-compromising, and reputation-undermining.In this article, you’ll learn why one should follow the medical device software requirements, what they are, and why you as a developer should think about it before planning for healthcare IoT development.
Why Complying With Medical Device Software Requirements Is a Must?
Medical equipment software requirements mean the policies and standards that are adopted by the healthcare market regulation authorities to ensure the safety of the device or its software. With the rise of the medical IoT and its annual market size growth, the need to regulate its development is motivated by the potential security and safety challenges.
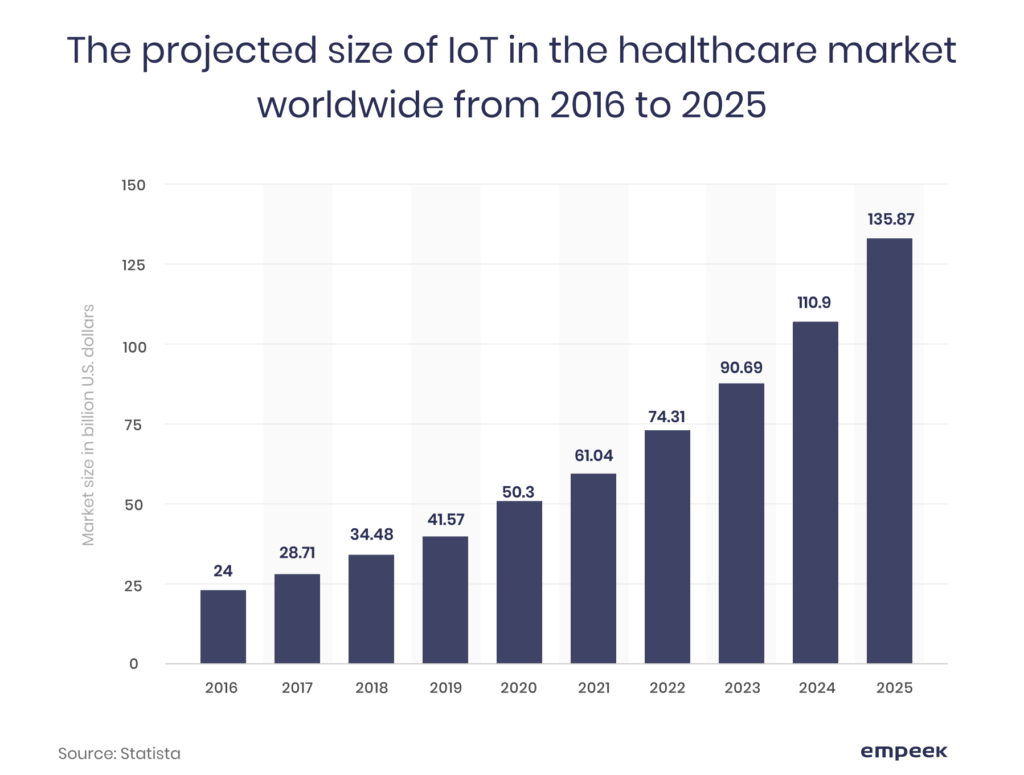
Depending on the market, these regulations and regulatory agencies may vary, though there are some adopted standards for medical device compliance we’re going to talk about in a bit. Now, let’s see why complying with their requirements is a must for a software developer.
1. To Ensure Patients’ Safety
Cause no harm is the first principle of medicine, and it applies to medical iot solutions and services as well. Patient’s safety and the security of their information is your primary guide during any stage of development. But even if you apply the best industry practices and technological novelties, how can you be sure that your creation is safe? This is where the regulations come in handy. With the set standards for medical device compliance and software requirements, you can be confident that your solution won’t compromise any aspect of the patient’s safety and wellbeing.

2. To Get Approved by the Regulator
If the patients using your IoT are safe, it means that your IoT can be approved by the regulator of the market you want to supply it for. Safety and Quality are the two primary focuses of any regulatory authority regardless of the market. However, depending on the country where you want your solution to be adopted, you need to obtain approval stamps from its regulators. For the USA, you need to follow the FDA medical device software guidance, and to market the software worldwide, you need to follow the ISO IEC 62304 standards. Usually, you should have both approvals to avoid penalties in case your creation may challenge the patient’s safety and to enter the market at all.
3. To Avert Medical Device Compliance Problems During an Audit
The logic here is simple. If you’ve developed your SaaS or software as a medical device (SaMD) according to the adopted standard for medical device software in the particular market, and have a well-documented traceability matrix, you’re ready for any audit at any time. Usually, such audits take place annually and their verdict can either allow you to either continue producing and market your device or face penalties. The latter can be in the form of fines, recalls, and reputational damage that entails.
4. To Prevent Negative Feedback from the Clients
Complying with the guidelines after you’ve developed the product for your client is a surefire way to mess up the deadline. Why? Because chances are, you’ll have to make it all over again, and for the clients, it means they’ll have to wait. This most likely will be mentioned near the 3-star rate they will leave on your website.
These are the main reasons to accelerate compliance with the medical IoT industry standards. Let’s see what are their main requirements.
Standard for Medical Device Software
The medical industry is strictly regulated worldwide, and to make it to market, your creation usually has to comply with many issues. Regulating the medical software and software as medical devices happens within the scope of regulation of the medical devices themselves, and they all depend on the medical device class regarding the potential harm to the patient. To such belong:
- EU Medical Device Regulation (for EU market)
- FDA Regulation (for U.S. market)
- ISO IEC 62304 regulation (globally accepted standard)
Let’s review each of these standards in more detail.
EU Medical Device Regulation
If the software falls under a medical device software (MDSW) category, it should comply with the requirements of Medical Device Regulation (former Medical Devices Directive). Here is an algorithm that helps to define whether your creation is covered by these regulations.
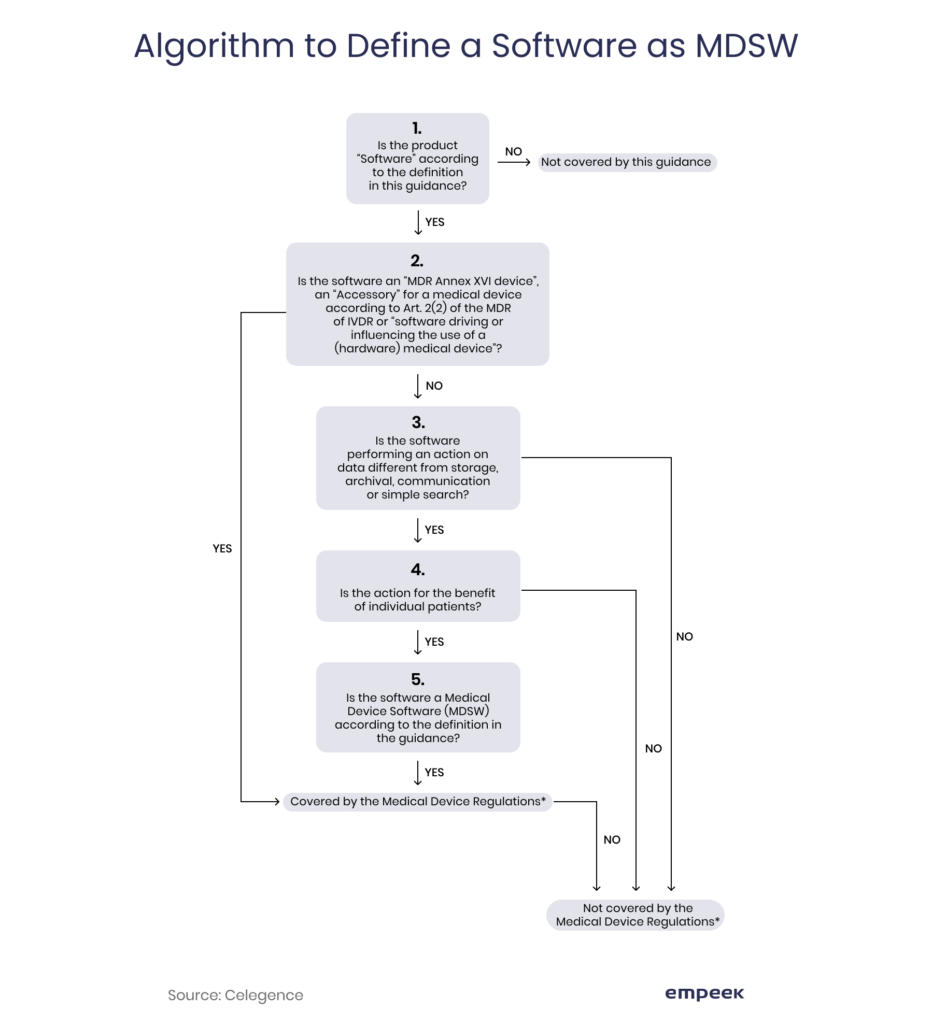
An MDSW is software that:
- Directly controls a medical device (hardware) and provides immediate decision-triggering information intended to be used by healthcare professionals or patients (e.g., blood glucose meter software)
- Provides support for healthcare professionals (e.g., ECG interpretation software).
- Process, analyze, create, or modify medical information when the software is governed by a medical intended purpose (e.g., software that assesses the scans to detect the pathology and prove the clinician’s hypothesis)
- Has its intended medical purpose.
All the MDSWs are assessed by the risk factors and can be classified as Class I, II, or II MDSW. Regardless of what class the software falls into, you should do these steps to get the approval sign:
- Use Quality Management System (QMS), the framework for which is defined in ISO:13485
- Conduct clinical evaluation, i.e. Post-Market Clinical Follow Up (PMCF)
- Provide all technical documentation with information on device description and specification + its variants and accessories, information to be supplied by the manufacturer, design and manufacturing information, general safety and performance requirements, risk-benefit analysis and risk management, and product verification and validation.

FDA Regulation
The FDA regulation considers 3 types of software used in healthcare:
- Software as Medical Device (SaMD), which is a medical device on its own
- Software in a Medical Device (SiMD), which is essential for the device to function
- Software, which is used in the manufacture or maintenance of a medical device.
The regulation of the software you develop depends on what class of medical equipment your software supports.
The main document that sets the requirements for FDA medical device software approval is Title 21 “Food and Drugs” of the Code of Federal Regulations. Special attention should be paid to the regulation regarding:
- Electronic Records (21 CFR Part 11)
- Establishment Registration and Medical Device Listing (21 CFR Part 807)
- Pre-Market Notification 510(k) (21 CFR Part 807 Subpart E)
- Pre-Market Approval Application (21 CFR Part 814)
- Investigational Device Exemption (21 CFR Part 812)
- Quality System Regulations (21 CFR Part 820)
- Labeling (21 CFR Part 801)
- Medical Device Reporting (21 CFR Part 803)
ISO IEC 62304
Complying with internationally adopted standard IEC 62304 is a must for the medical software developers who enter the international market. These compliance standards apply when the software is a medical device itself (SaMD) or functions as the device’s integral part (SiMD). According to the potential damage level, the software is classified as:
- Class A: No injury/damage to health may occur
- Class B: Not serious injury may occur
- Class C: Death or serious injury may occur
The ISO framework sets the needed processes, tasks, and activities that the developers need to follow to ensure the compliant software life cycle processes.
In parts 5-9 of the IEC 62304 define the main regulatory issues and guidelines. They concern:
- Software development process
- Software maintenance process
- Risk management
- Configuration management
- Problem resolution process
Any regulation needs detailed processing and studying to make sure the development process and the final product’s lifecycle comply with all the requirements. Empeek prepared a few tips to simplify this process for you.
Tips to Ensure Medical Device Compliance
- Understand the regulatory requirements of the market you design the software for and base your development process on following each of them.
- Create a traceability matrix that allows you to prove compliance with the regulatory requirements for medical devices.
- Choose electronic quality management systems. These solutions will help you to keep track of all development stages, document and index all the changes and technical documentation, ensure an agile approach while remaining easily traceable during the audits, record quality processes, allow phase gating, enable communication, and even make audits possible within a click.
- Run tests often and document them.
- Invite third-party audit companies to assess the compliance for medical device software in the target market.
Final Thoughts
Medical device software compliance standards are important for the developers to get a chance to market their solutions worldwide. For healthcare providers, it’s a must to stay market-competitive, provide safe services to their patients, and avoid possible fines for adopting non-compliant or disruptive technologies. Empeek developers base the development process on the regulatory guidelines to ensure that the solution will meet all the industry and market standards from the beginning. Let’s discuss how our software can benefit your practice today!



